

在生物制药工艺中,过滤是确保产品无菌性、澄清度和安全性的关键单元操作。滤器(包括滤膜和壳体)与药液的直接接触,使其成为潜在的污染源。其中,“相容性风险浸出物”是指在特定工艺条件下(如pH、温度、溶剂、时间),从滤器材料中迁移至药液中的化学物质。这些浸出物可能影响药物的稳定性、安全性(如引发毒性或免疫反

HPLC和UPLC对比表特性HPLC(高效液相色谱)UPLC(超高效液相色谱)带来的优势工作压力较低,通常≤ 400 bar极高,通常600-1500 bar允许使用更小颗粒的填料和更快的流速色谱柱填料粒径较大,通常3-5 μm更小,通常1.7-2 μm更高的柱效,分离能力更强,峰更尖锐对称分析速度较慢非常快 (比HPLC快3-5倍)节省时间,提高样品通

在药物研发、天然产物提取等尖端科学领域,科学家们同样面临着从复杂混合物中“淘”出目标珍宝的挑战——这一过程,就是制备液相色谱技术所成就的现代“化学炼金术”。制备液相色谱的核心原理亦是基于混合物中各组分在固定相与流动相之间作用力的差异,从而实现顺序洗脱、分离提纯。二者本质都是“利用差异实现选择性富集

体外释放试验(in vitro release test,IVRT)是表征和评价半固体制剂性能的有效手段,体外释放速率可以反映药物的溶解度、粒径、剂型流变性等多种理化参数的综合作用,可辨别处方和工艺变化对制剂的影响,是产品开发、质量控制、稳定性考察及产品批准后变更的重要质量控制项目。&nbs
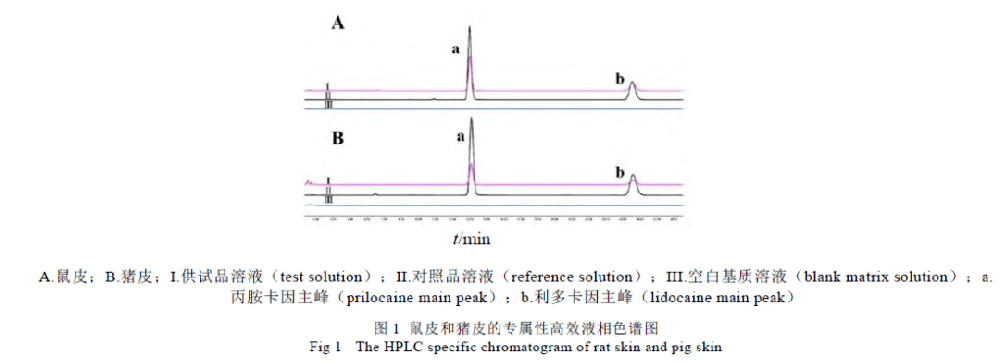
目的:综合国内外关于半固体制剂的体外药物释放试验的研究,探讨关于体外药物释放试验各装置特点及试验条件的选择并判断释药模型。方法:根据国内外31篇文献,对如何根据所研究半固体制剂的特点选择合适装置及试验条件进行体外药物释放试验进行综述。结果:通过药物的体外释放结果判断出药物符合的释药模型并阐明释药机理。
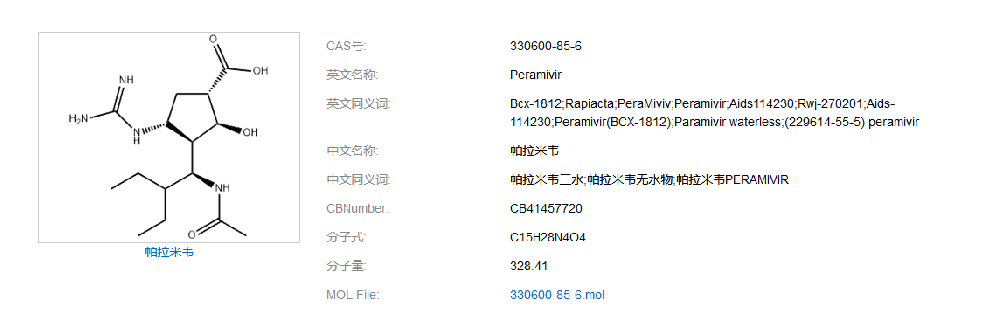
如何建立帕拉米韦质量标准,现在百度检索:同时在药物在线(https://www.drugfuture.com/standard/):各国药典均未收录载帕拉米韦。无药典收载、无进口标准:医学百科(https://www.yixue.com):帕拉米韦水合物是美国BioCryst 公司开发的以流感病毒表面糖蛋白神经氨酸酶为作用靶点的新型环戊烷类抗流感病毒制剂,是世界首

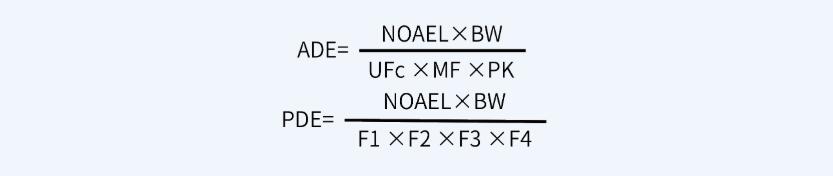
ICH Q3E原文:“虽然在此类情况下,可以接受短期暴露的毒性研究作为 PoD,但这不包括 LD50 研究。”出自ICH Q3E指南关于毒性评估中关键调整因子F3(研究持续时间因子)的阐述部分。这句话虽然简短,但蕴含了ICH Q3E基于风险、科学为本的评估理念,以及对传统毒理学方法局限性的明确界定。以下将

2018年,“沙坦类药物检出NDMA"事件震惊全球制药行业,引发FDA对亚硝胺类杂质的系统性排查,目前全球已有超20个国家发布针对性法规,其中FDA的《Controlof Nitrosamine lmpurities in HumanDrugs》(2024年修订版)与《Recommended Acceptable Intake Limits for NDSRls》(2023年)两份指南,构建了当前最严格的亚硝

变更相关问题问题1:附条件批准上市药品在二期沟通时,提出在上市申请审评期间做生产厂地的变更,请问是否可行?答:首先我们不建议在上市审评期间做生产厂地变更。不管是一类药、二类药、三类药还是四类药,确实在审评期间有一个提交变更的通道,但根据《药品注册管理办法》,审评期间发生重大变更时,上市申请是要撤
药品中亚硝胺的潜在风险始终是全球医药行业关注的焦点。我们在开展药品亚硝胺风险的评估时,往往关注于原料的引入情况以及制剂的降解,而对于辅料的影响却关注不足。其实,辅料对亚硝胺形成的潜在影响不容忽视。尽管辅料直接引入亚硝胺的风险极低,但其所含的特定反应性物质(如亚硝化剂或胺类成分)可能间接促进或抑制亚硝
亚硝胺英文来源药物名称AI值(ng/day)CPCA潜在分类发表日期N-Methyl-N-nitrosophenethylamine(13256-11-6, NMPEA)-N-甲基-N-亚硝基甲胺85-Jan-233-((Ethyl(nitroso)amino)methyl) benzenesulfonateDextromethorphan右美沙芬18129-Jul-24N-Nitroso-2,4-thiazole amine /N-((2-isopropylthiazol-4-yl)methyl)-N-methylnitrous

1 国内外遗传毒性杂质监管现状1 从宏观上解读杂质1.1 杂质与药物不良反应的关系很多同仁都认为杂质与药物的不良反应息息相关,认为杂质越小或越少、临床不良反应发生几率也就越小或越少,进而在进行杂质研究与控制时,力求面面俱到、尽善尽美。殊不知,引起药物不良反应的原因是多方面的,并不仅仅是药物中的杂质。人用药品
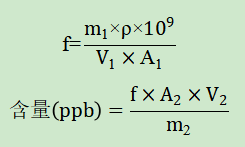
一、验证目的本验证方案的目的是为了判断所采用分析方法是否科学、合理,能否有效的检测盐酸度洛西汀中基因毒性杂质N-亚硝基二甲胺(NDMA)和1-氟萘的含量。根据客户要求,N-亚硝基二甲胺(NDMA)限度为0.8ppm,1-氟萘限度为12.5ppm。根据此限度要求,对盐酸度洛西汀中NDMA和1-氟萘的检测方法进行验证,验证内容包括系统适用
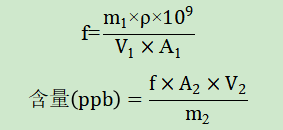
一、验证目的本验证方案的目的是为了判断所采用分析方法是否科学、合理,能否有效的检测头孢地尼中N-亚硝基二甲胺(NDMA)和N-亚硝基二乙胺(NDEA)的含量。参考头孢地尼胶囊的说明书,每日最大用量为0.3g,根据FDA发布的《人用药中亚硝胺杂质控制》行业指南,NDMA和NDEA每日可接受摄入量分别为:96.0ng/天和26.5ng/天,计算

一、研究目的包装系统是指容纳和保护药品的所有组件,包括直接接触药品的包装组件和起额外保护作用的次级包装组件。作为药品包装组件,一方面应满足系统对密封性的要求,为药品提供保护并符合包装预期使用功能;另一方面还应与药品具有良好的相容性,即不可引入存在安全风险的浸出物,或浸出物水平符合安全性要求,且不会因
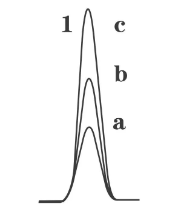
我们药物杂质分析方法的开发的时候,经常会遇到分析物中包含了较高浓度的主成分(也就是药物活性成分)和很低浓度的杂质(包括工艺杂质及降解杂质)。而为了兼顾低浓度杂质的灵敏度,必然要增大进样体积,或者提高进样浓度,确保定量的准确性。因此,某些时候就会造成色谱柱的过载。那么色谱柱的过载又可以分为质量过载和体

一、研究目的包装系统是指容纳和保护药品的所有组件,包括直接接触药品的包装组件和起额外保护作用的次级包装组件。作为药品包装组件,一方面应满足系统对密封性的要求,为药品提供保护并符合包装预期使用功能;另一方面还应与药品具有良好的相容性,即不可引入存在安全风险的浸出物,或浸出物水平符合安全性要求,且不会因
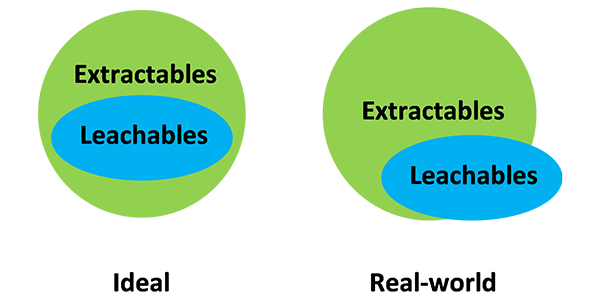
01可提取物与浸出物(E&L)研究概述可提取物(Extractables):在更具侵蚀性(More aggressive)条件下(例如模型溶剂、高温、高离子强度、恶劣pH、高接触时间等),可以从工艺系统的组件接触表面提取出的化学物质。尽管在生物制药生产过程中不经常遇到这种侵蚀性条件,但可提取物仍然很重要,因为有关可提取物的知识可以帮助

各种不同的分析、分离纯化和结构鉴定技术应用于药物研发的整体过程,其中对化合物的纯度确定是分析工作的重要内容之一。在新药研发过程中,许多待标定的化合物是新化合物,没有已知纯度的自身标准品。这时,需要经验丰富的分析人员对化合物进行各种不同的测试,主要包括HPLC纯度、有关物质、杂质、水分、残留溶剂、无机盐等
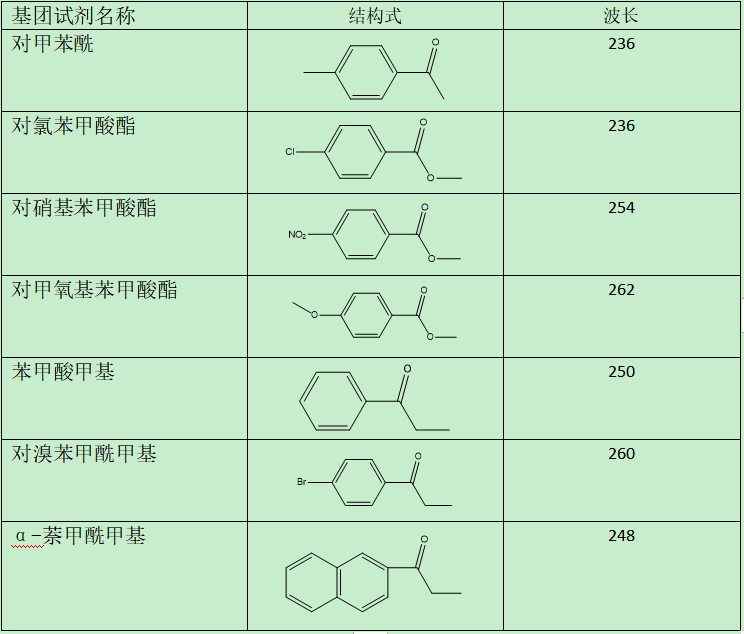
杂质没有紫外吸收怎么办? 传统化学方法,无法准确定量,怎么办? 示差检测器,蒸发光检测器,灵敏度低,满足不了检测限要求,怎么办? LC-MS,价格昂贵,适用范围窄,怎么办? 在整个药品研发过程中,产品杂质研究是至关重要的一环,一些特殊杂质

起始原料(Starting material)是构成API 结构的重要结构组成部分的一种原料、中间体或API。它可以是已上市的商品、以合同或商业协议方式购自一家或多家供应商的产品,或是企业自己生产的物质。根据国内外GMP要求,起始原料作为GMP监控的起点,需要接受国内外官方机构的监督检查。❖ 起始原料选择依据依据ICH Q

为证明所采用的分析方法是否适合于相应检测要求和目的,被测样品质量是否可控,保证得到一致、可靠和准确的测定结果,检测人员是否有能力操作分析方法,进行化学药品分析方法的验证、转移和确认。 方法验证(method validation)[概念]实验室通过试验设计和测试,对方法学
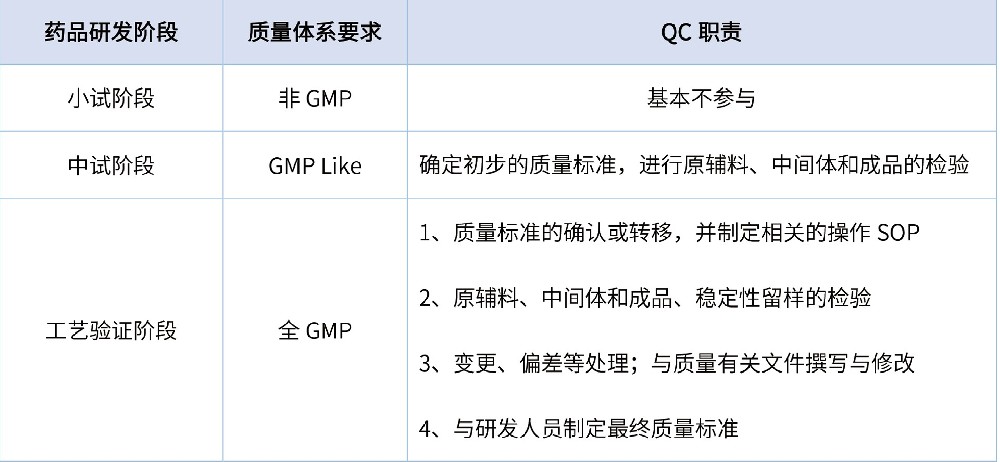
按照最新版《药品管理法》(2020版)要求,在药品全生命周期中需执行四个最严,即“最严谨的标准、最严格的监管、最严厉的处罚、最严肃的问责”。为保证药品的安全性和有效性,药品研发作为药品生命周期的源头,需在遵循《药品管理法》的前提下执行好《药品注册管理办法》法规要求。药品研发按创新药、改良型新药、仿制药进
药品生产后必须对其生产设备采取一定的程序进行清洗,防止药物成分残留到下一批产品中,形成交叉污染,从而影响下一批产品的质量及安全性。清洁验证是证实清洗程序合理性的必要技术手段。WHO、EU、ICH、FDA及我国药品GMP均对药品生产清洁验证提出了相关要求:1)使用同一生
导读:近期CDE官网更新了:仿制药质量和疗效一致性评价百问百答(第3期),其中包括「杂质研究+溶出曲线对比」新规问:口服固体制剂一致性评价杂质研究需要注意哪些问题?答:口服固体一致性评价品种均为已上市品种,杂质研究中可以参考的文献资料较多,但是杂质研究不全面或过度研究,均可能给研发和审评带来不必要的负担,
对于药物分析的工作人员来说,检验方法的验证和确认步骤是必须了解的知识。本文对检验方法的验证和确认步骤及详细计算方法进行整理,供各位同仁学习参考。一、检验方法验证的基本内容检验方法验证的基本内容包括方案的起草及审批,检测仪器的确认。适用性验证(包括准确度试验、精密度测定、线性范围试验

一、概述辅料一般被看作是惰性物质,但是当辅料被添加到制剂中时,某些辅料可能会对药物的最终药理学作用产生非常大的影响。药物和辅料之间的相互作用可能是物理化学作用,也可能是生理作用,对制剂的稳定性、安全性和有效性均有一定程度的影响。其相互作用大小与药物的性质、辅料的性质及其加入量均有较大的关系,其相互作

1、柱容量柱容量越大越不容易过载。决定柱容量的指标是柱截面上固定相的面积占比,它与空容、填料的比表面积、表面健合率等色谱参数有关。需要注意的是,增加柱长并不能增加柱容量,因为样品进样后不可能立即分布于整个色谱柱,同样因为无限直径效应的缘故,增大柱内径也不能增大柱容量。2、进样量显然,减小进样量可



 在线客服
在线客服
 快速发布
快速发布
 文章查询
文章查询
 分析化学
分析化学
 我的消息
我的消息
 文章发表
文章发表

 回到顶部
回到顶部