
化学分析网 www.chemanaly.com根据国家相关法律法规要求,药品上市许可持有人(申请人)应当主动开展药品上市后研究,实现药品全生命周期管理。鼓励持有人运用新生产技术、新方法、新设备、新科技成果,不断改进和优化生产工艺,持续提高药品质量,提升药品安全性、有效性和质量可控性。但药品上市后变更不得对药品的安全性、有效性和质量可控性产生不良影响。

药品某一项变更往往不是独立发生的,一项变更伴随或引发其他变更称之为关联变更。由于这些变更对药品安全性、有效性和质量可控性影响程度可能不同,研究工作总体上应按照技术要求较高的变更类别进行,同时建议关注多项关联变更对药品安全性、有效性和质量可控性产生的叠加影响。
作者此文重点对化学原料药变更的分类以及研究要求进行梳理,方便大家对比学习,赶快收藏起来吧!
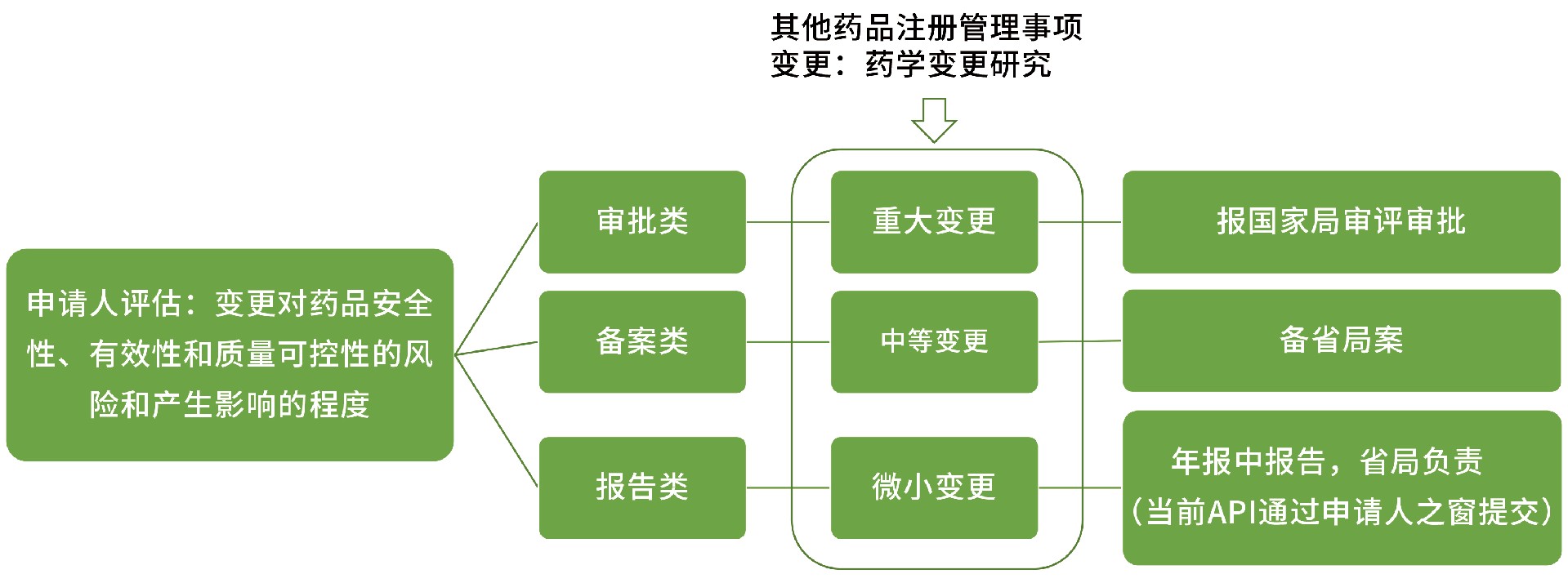
药品上市后变更包括注册管理事项变更和生产监管事项变更,也就是涉及注册批件和生产许可证相关事项的变更。依据变更对药品安全性、有效性和质量可控性的风险和产生影响的程度,将变更划分为审评类变更,备案类变更和报告类变更。

我们常说的药学变更研究,一般都是针对注册管理事项发起的变更。本文对原料药的药学变更分类和研究进行简单的介绍。
根据不同时间阶段分类:
上市后变更:参照《药品上市后变更管理办法(试行)》、《已上市化学药品药学变更研究技术指导原则(试行)》进行研究。
化学分析网 www.chemanaly.com
审评期间变更:参照《药品注册申请审评期间变更工作程序》,审评期间允许进行变更申请,这也是近期变更研究的热门话题,审评期间的变更需要注意以下方面:
不影响产品质量
仅能提出一次
参照已上市药品变更相关指导原则研究及申请
时限按变更/原申报剩余时间最长计算
审评期间的变更以补充申请报CDE
状态为I,未进入审评的原料药变更:可参照已上市药品变更研究,当前原料药受理后即视为启动审评,暂时没有途径递交资料,后续CDE应该会有新的政策出来。
根据变更对药品的影响程度分类: 化学分析网 www.chemanaly.com
依据变更对药品安全性、有效性、质量可控性的影响,将药学变更分为微小变更、中等变更和重大变更,由申请人依据对产品的理解进行评估及相应的研究、申报工作。
变更研究
微小变更:
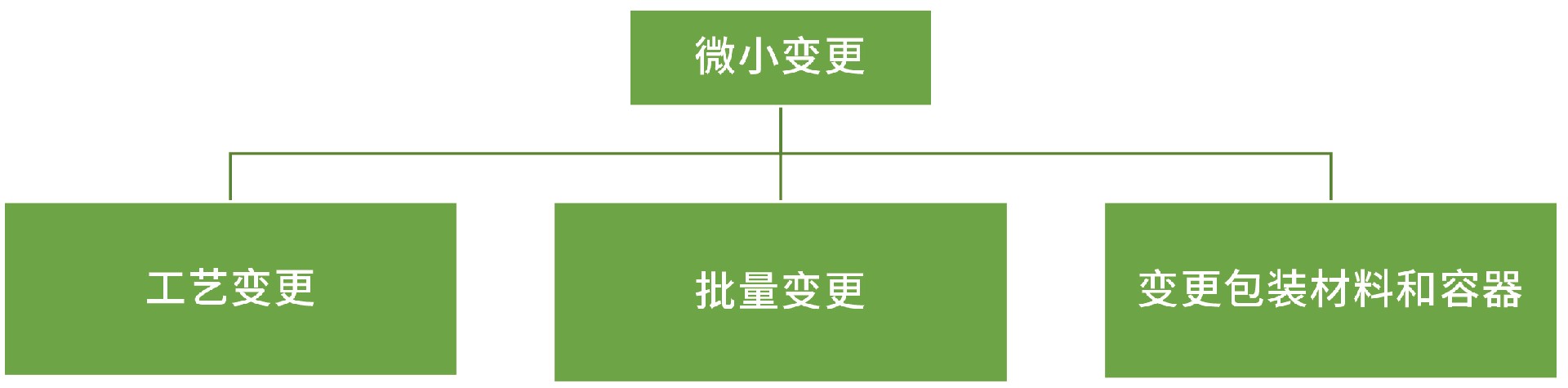
工艺变更:
增加/收严过程控制;
提高起始原料、中间体的质量标准;
变更试剂、溶剂的质量标准或级别,但不降低质量;
变更最后一步反应之前生产设备;
变更最后一步生产设备,且材质、设计和工作原理不变,原料药关键质量不变;
变更起始原料的供应商,起始原料的合成路线不变,且起始原料的质量不降低。
研究内容:
变更情况及原因;
一批样品检验;
年报中报告首批样品的长期稳定性试验数据。
批量变更:生产批量变更在原批准批量的10倍以内(包括10倍)
研究内容:
设备对比;
一批批生产记录;
变更前后质量对比研究;
1-3 批样品进行检验。
变更包装材料和容器:
变更原料药装量;
固体原料药的包材(包材为A)。
研究内容:变更前后包材对比。
中等变更:
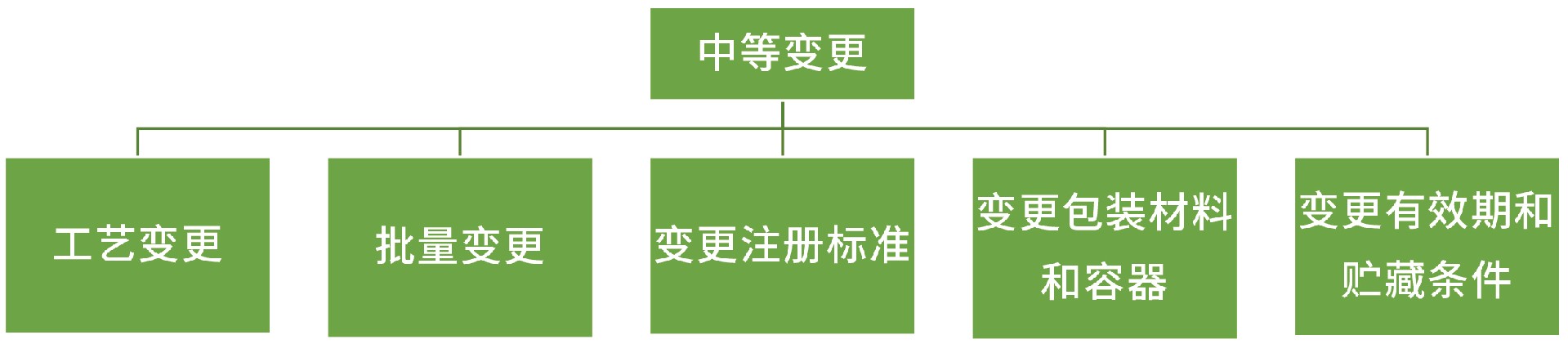
工艺变更:
延长工艺路线,且延长的工艺路线与原起始原料一致;
变更起始原料的合成路线,起始原料的质量不降低;
变更最后一步反应之前的工艺步骤中的反应试剂、溶剂种类、生产条件等,但原料药杂质谱保持一致;
将返工工艺作为固定的生产步骤纳入注册生产工艺;
变更起始原料、中间体的质量标准,变更后起始原料、中间体的质量控制水平不得降低;
变更起始原料的供应商,起始原料的合成路线不变,且起始原料的质量不降低;
无菌原料药变更除菌过滤过程的滤过参数/除菌工艺过滤器从单一过滤器改为两个无菌级过滤器串联。
研究内容:
变更情况及原因;
一批批生产记录;
质量对比研究,变更前后样品的杂质谱等一致;
1-3 批样品进行检验;
一批样品加速及长期稳定性考察,提供 3 个月的稳定性研究资料。
批量变更:生产批量变更在原批准批量的10倍以上。
研究内容:设备及工艺对比。
变更注册标准:
新增检验项目;
在原标准规定范围内收紧限度;
注册标准中文字描述的变更。
研究内容:
对质量标准变更合理性进行研究;
不用提供生产记录及稳定性数据;
多批次检测数据。
变更包装材料和容器:无菌和/或液体原料药的包装材料和容器的材质和/或类型。
研究内容:
变更前后包材对比;
进行包装工艺验证(不要求全工艺验证)。
变更有效期和贮藏条件:
延长药品有效期;
缩短药品有效期。
研究内容:
变更情况及原因;
3 批样品长期稳定性数据。
重大变更:
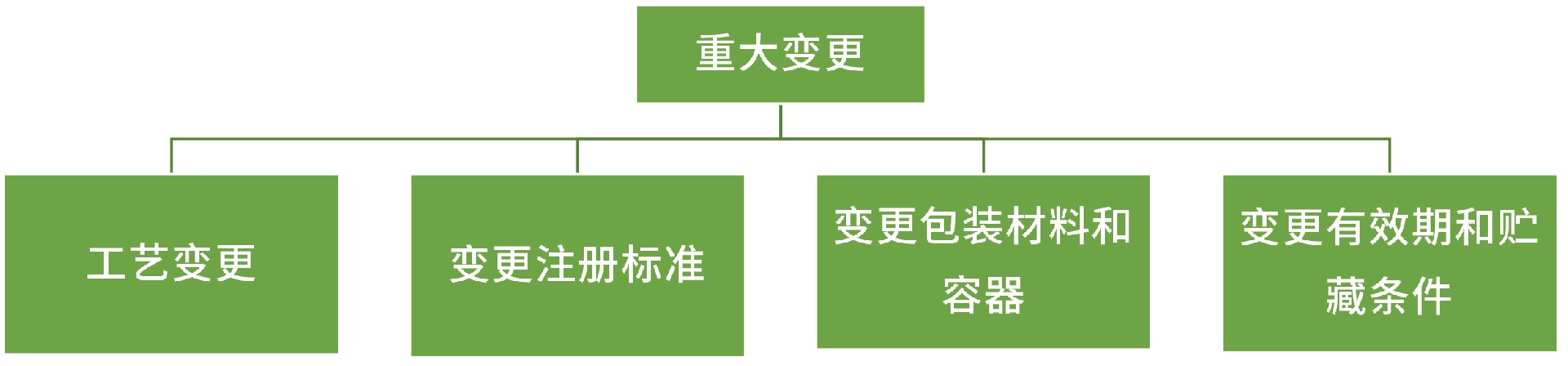
工艺变更:
变更原料药合成路线(除路线延长);
变更起始原料的合成路线,起始原料的质量发生变化;
变更最后一步反应及之后的生产工艺;
变更可能影响原料药关键质量属性的工艺参数;
在注册生产工艺中增加重新加工工艺;
放宽或删除已批准的起始原料、中间体质量控制和生产过程控制;
变更原料药生产工艺中的设备,可能导致原料药杂质谱或关键理化性质发生变化;
无菌原料药生产过程变更灭菌/无菌工艺,变更除菌过滤器孔径;
其他可能导致原料药杂质谱和关键理化性质与变更前不一致的变更。
研究内容:
变更情况及原因;
变更后的API和中间体结构确证;
一批批生产记录;
质量对比研究;
3 批样品检验;
三批样品加速及长期稳定性考察,3~6个月的稳定性对比研究。
变更注册标准:
变更检验方法;
放宽控制限度;
删除注册标准中的任何项目。
研究内容:
对质量标准变更合理性进行研究;
3批样品检测数据;
变更包装材料和容器:
变更为全新材料、全新结构、风险度提高的新用途的包装材料和容器;
变更纳入登记管理的包装材料和容器,变更后的包装材料和容器尚未登记或登记状态为I。
研究内容:
变更前后包材对比;
进行包装工艺验证(不要求全工艺验证)。
变更有效期和贮藏条件:
变更药品贮藏条件;
由于药品的其他变更导致的有效期变更。
研究内容:
变更情况及原因;
按方案,3 批样品稳定性,3-6月数据。
除以上变更外,《药品上市后变更管理办法(试行)》及《已上市化学药品药学变更研究技术指导原则(试行)》对持有人及生产场地的变更流程及研究内容进行了明确的规定。
持有人变更:
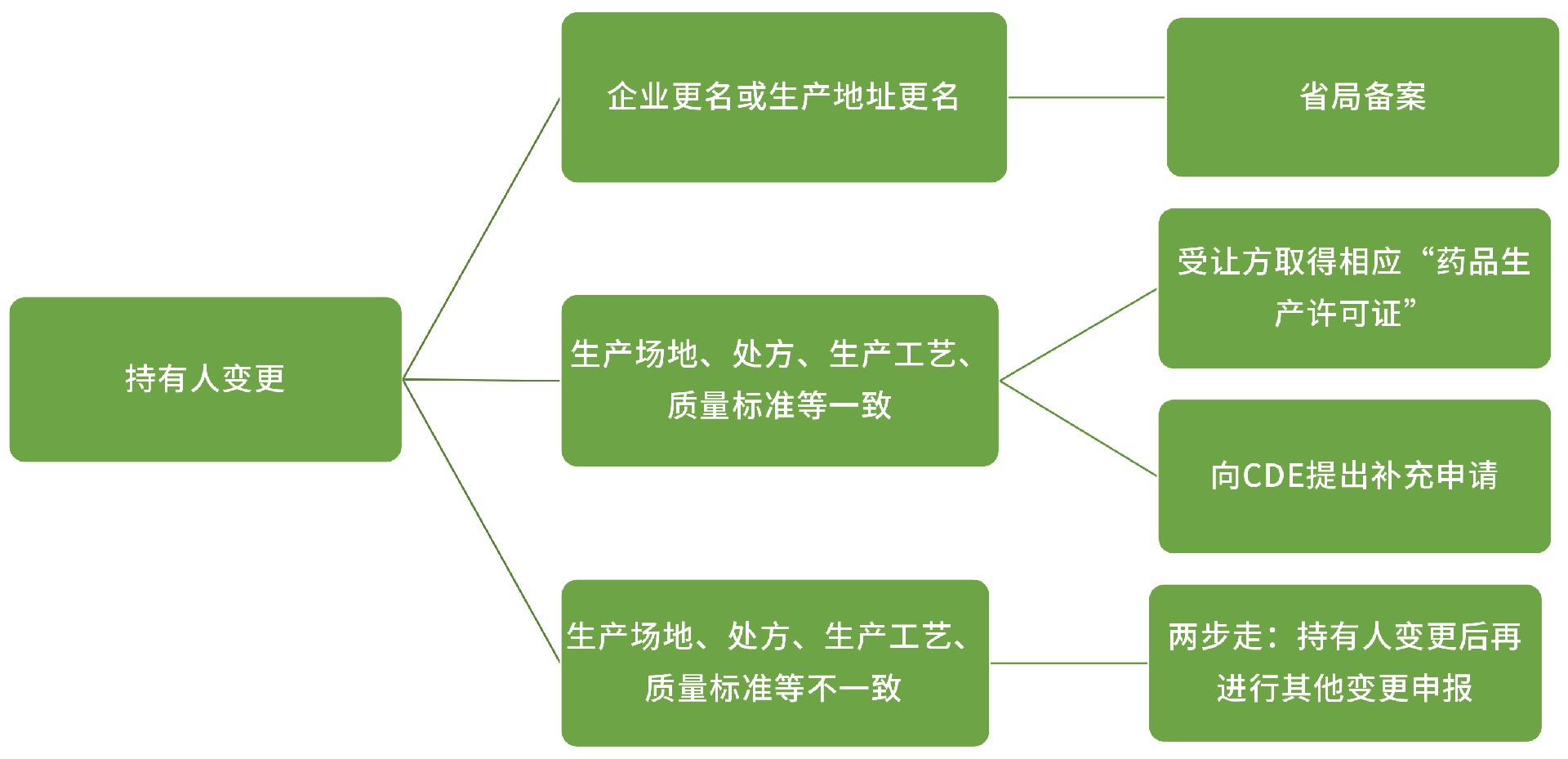
生产场地变更:
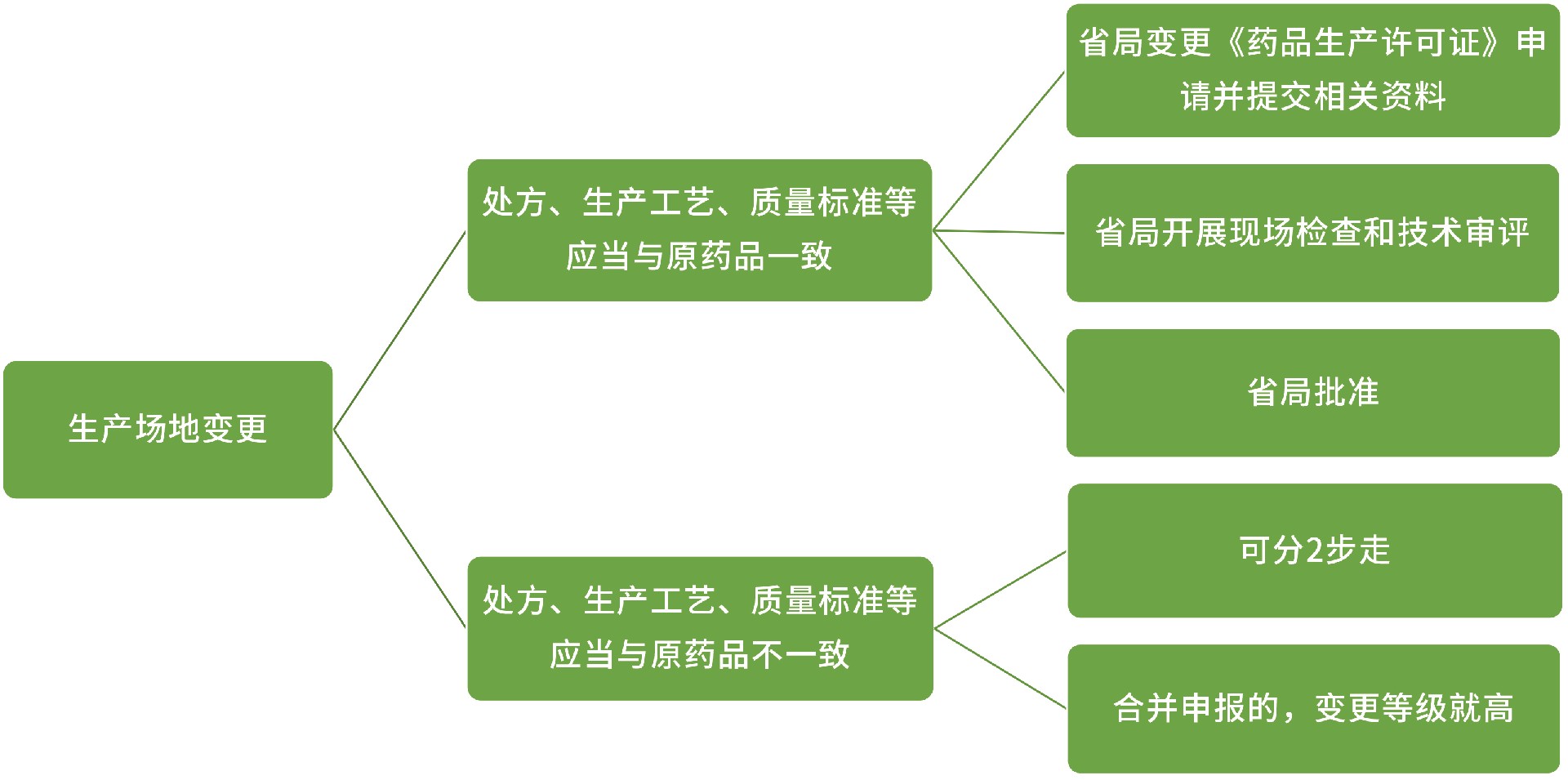
在同一生产地址内变更非无菌生产步骤:
研究内容:
变更的具体情况和原因;
比较新旧场地生产工艺情况(含设备和工艺);
对变更后一批样品进行检验,应符合质量标准规定;
对变更后首批样品进行长期稳定性考察,并在年报中报告。
上报:省局
在同一生产地址内变更无菌步骤 OR 生产地址变更至另一不同生产地址:
研究内容:
变更的具体情况和原因,进行验证;
比较新旧场地生产工艺情况(含设备和工艺);
变更后一批样品的批生产记录;
进行质量对比研究,关键理化性质和 杂质谱等应保持一致;
对变更后三批样品进行检验;
变更后一批样品进行加速及长期稳定性考察,提供3个月的稳定性研究资料,并与变更前产品比较,变更后样品的稳定性应不低于变更前。
上报:省局
案例分享
案例:某原料药,由于起始原料供应商不再供货,对生产工艺的进一步研究及制剂对原料药质量要求的提高,拟进行一系列变更:
起始原料变更:
工艺优化:
T项目注册标准变更:
解析:案例该项目涉及一系列变更,有属于微小变更的改变后处理溶剂,中等变更的收严限度,重大变更的变更最后一步反应参数等,依据
《已上市化学药品药学变更研究技术指导原则(试行)》,按照技术要求较高的类别进行研究,故本品按重大变更进行研究,报补充申请,并依据具体变更内容进行针对性研究。需要研究内容如下:
对变更前后的路线进行详细的对比说明
对变更前后的起始物料、中间体、原料药进行对比研究
进行工艺参数评估,拟定合理参数范围
对变更后生产的原料药进行结构确证
进行三批工艺验证,对所得中间体和原料药进行全面分析研究,提供3批检测报告
对变更后三批样品进行加速及长期稳定性考察,提供3-6个月的稳定性研究资料
变更流程及时限
备注:自备案完成之日起30日内完成对备案资料的审查,必要时可实施检查与检验;审批类变更的补充申请审评时限为六十日;补充申请合并申报事项的,审评时限为八十日;其中涉及临床试验研究数据审查、药品注册核查检验的审评时限为二百日。
主要法律依据:
《中华人民共和国药品管理法》.2019.8
《药品生产质量管理规范(2010年修订)》.2010.10
《药品注册管理办法》.2020.3
《药品上市后变更管理办法(试行)》.2021.1
《已上市化学药品药学变更研究技术指导原则(试行)》.2021.2
《已上市化学药品变更事项及申报资料要求》.2021.2
《药品生产场地变更研究技术指导原则(征求意见稿)》.2017.10
《药品注册申请审评期间变更工作程序》.2021.6
创新药(化学药)临床试验期间药学变更技术指导原则(试行).2021.3
免责声明:本站提供的一切文章和内容信息仅限用于学习和研究目的;不得将上述内容用于商业或者非法用途,否则,一切后果请用户自负。本站信息来自网络收集整理,版权争议与本站无关。我们非常重视版权问题,如有侵权请邮件与我们联系处理。敬请谅解!
本文最后更新于2022-11-28 22:00:13,如果你的问题还没有解决,可以加入交流群和群友们一起讨论。


 在线客服
在线客服
 快速发布
快速发布
 文章查询
文章查询
 分析化学
分析化学
 我的消息
我的消息
 文章发表
文章发表

 回到顶部
回到顶部